










CrystalMaker X 10.6.3 macOS | 317 MB
CrystalMaker X is the most-efficient way to visualize crystal and molecular structures. Its interactive design lets you "see the wood for the trees" and build your own visual understanding of complex materials. CrystalMaker transcends traditional crystallography software, letting you create dynamical visualizations with rotatable animations. Energy-modelling tools let you design and relax new structures, predict their vibrational properties, and explore how they interface with other materials.
CrystalMaker provides a streamlined workflow that majors on productivity: just drag-and-drop your data files into the program for instant display in spectacular photo-realistic colour. Manipulate structures in real time, with the mouse. Multiple View "bookmarks" and undo levels encourage exploration and discovery - ideal for teaching and research.
Integrated Structures Library - Now Add Your Own
CrystalMaker X includes an integrated structures library with some 1200 structures: annotated, indexed and with previews - ready for immediate display. The library includes over 400 minerals (all the the major rock-forming minerals plus many more), as well as important inorganic and organic crystals and molecules: from buckyballs to zeolites, and from dental ceramics to high-Tc superconductors. Also included is a teaching library of crystal-chemical type structures, plus thematic libraries covering topics as diverse as chemical warfare, medicinal chemistry, food flavourings and more.
Not Just Another Library...
These aren't your typical crude CIF text files! All files are saved using CrystalMaker's rich naixava document format (CMDX or CMDF) which lets you instantly load a fully-completed model with bonding, polyhedra, detailed notes (with publication data in most cases) and full metadata for powerful searching.
Each structure has been carefully constructed, with the optimum view direction and model settings to convey the best-possible visual understanding - and thereby saving you valuable time.
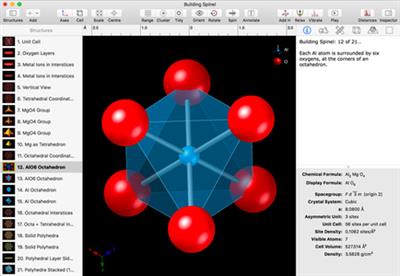
Many crystal files include multiple structure views, to showcase different aspects of the structure (e.g., a complex framework broken down into its consistuent building blocks, such as (Al,Si)O4 tetrahedra, rings of tetrahedra, cages, channels and building up a full network. You can use these files as teaching resources: just open the file, hit the Play button, and discover a whole new world of nano-scale beauty.
You can add an of these structures to an existing document window, for you to edit or share with colleagues and students.
You can browse structures in the Library's Browser view: grouping by folder, or flat view or simply exploring rotating structures and their notes in the Library's integrated "CrystalViewer". This isn't just a static library: you can add your own content, grouping structures into folders and taking advantage of CrystalMaker's powerful search capabilities: search by filename, notes or chemical formula.
Comprehensive Data Import & Export
Whilst the built-in libraries are great for teaching, researchers will want to visualize their own data - and they can! CrystalMaker X lets you import data from over 40 different formats: with instant display and powerful customization. CrystalMaker can handle including multi-structure files such as DL_POLY HISTORY - use CrystalMaker's synchronization and animation capabilities to rapidly understand structural behaviour, lattice dynamics, or visualize the trajectory of a simulation. CrystalMaker X can also handle truly massive structures. Take advantage of our unique "Depth Profiling" tool, to rapidly scan ares of interest in massive structures - ideal for characterizing the results from computer models.
3ED Volumetric
ATOMS (v4, v5)
CASTEP Cell
CASTEP Output
CASTEP Volumetric
CCL
Chem3D Cartesian
CIF
CMDX (CrystalMaker X Document)
CMDF (CrystalMaker 7-9 Crystal)
CMMF (CrystalMaker 7-9 Molecule)
CMTX (CrystalMaker Text)
CSSR
Gaussian CUBE
DEN (Volumetric)
DL_POLY Config
DL_POLY Revcon
DL_POLY History
DMol3 "CAR" file
DMol3 "ARC" trajectory file
FDAT (CCSD)
GRD (Volumetric)
GROMACS
GSAS
GULP
ICSD
LAMMPS dump
Molfile
PDB
RMCProfile
SDFile
SHELX
STRUPLO
SYSTRE
TOPAS
VASP Structure (POSCAR, CONTCAR)
VASP Trajectory (XDATCAR)
VASP Volumetric (CHG, CHGCAR, etc.)
VESTA
Voxel
WIEN2k Structure
XCrySDen Structure (XSF)
XCrySDen Animation (AXSF)
XYZ
A range of output options is available for sharing data with other programs, saving structural data, bond distances, coordination environments - or even building web pages with your data.
CASTEP Cell
Chem3D Cartesian
CIF
CMDX (CrystalMaker X Document)
CMTX (CrystalMaker Text)
Coordinates file
Visible Coordination file
Crystal Coordination file
Distances & Angles
DL_POLY Config
DL_POLY Revcon
DMol3 "CAR" file
Elements file
GRD volumetric
HTML Structure
PDB
RMCProfile
VASP Structure
Vibrational frequences & eigenvectors
Vibrational spectrum (IR)
Voxel volumetric
XYZ
Build New Crystals & Molecules Fast!
With CrystalMaker you can build any kind of crystal or molecular structure - quickly and easily. Built-in symmetry handling and the elegant spacegroup browser takes the slog out of crystallography, and the program will automatically generate all your bonds and polyhedra. Molecule building is even easier: point-and-click to add atoms and bonds. Use the new Relax command to minimize naixava energy and optimize your structure.
CrystalMaker provides a wide range of model types, including traditional "ball-and-stick", space-filling, polyhedral, wireframe and thermal ellipsoids. Each model type can be extensively customized, with the option of photo-realistic graphics or simpler, line-art display. CrystalMaker allows continuous Description range settings for millions of atoms, bonds and polyhedra.
Outstanding 3D Graphics
CrystalMaker X features industry-leading 3D graphics, for spectacular - and fast - pixel-perfect visualizations with perfect overlap correction and translucency. Enjoy high-resolution "Retina" graphics on the latest Macs or Surface Pro devices. Save publication-quality graphics - with transparency - or generate engaging videos of rotating structures or animations.
Real-Time Manipulation
Click-and-drag with the mouse, use the keyboard, or toolbar. View parallel to a vector or plane normal. Take advantage of multi-touch rotation and scaling with a trackpad, or simple hand gestures in space using a Leap Motion Controller (Windows version). CrystalMaker for Mac - as a proper native Mac application - sports a dedicated Touch Bar interface for the new MacBook Pro: rotate and zoom with dedicated touch dials, direction presets and sliders.
Multi-Structure Animation
CrystalMaker is the first program of its kind to go beyond static structures, to let you explore structural behaviour quickly and easily. Just drag-and-drop files into the same window, then rearrange their thumbnails to build your movie timeline. Manipulate and synchronize views, drag-and-drop thumbnails to rearrange them, then click the Play button and visualize the animation. It doesn't stop there: CrystalMaker X lets you rotate and scale animations, change their model types, view directions and model settings. You can also use the Playback Controls to "scrub" through an animation sequence: it's an ideal way to rapidly browse a simulation trajectory. Finally, you can export your animation as a movie - for use on the web or in a presentation.
Create Compelling Education Content
CrystalMaker documents can save multiple structures, each with its own graphical thumbnail. Whilst an obvious application is for structural behaviour and animation, you can also save different views of the same structure, different model types, atom ranges, or related structures. Since each structure can have its own notes, this makes an ideal educational tool - a full-interactive slideshow with rotatable 3D models, presented in context. Examples of how to build engaging educational content are included with the program (e.g., "Building Quartz", "Building Spinel").
Volumetric Data Visualization
CrystalMaker X lets you combine any number of 3D datasets with the current structure, each with its own visual settings. You can visualize data imported from text files, or generated within the program in electron density or porosity calculations.
Visualize volumetric data from 3ED, CASTEP, Gaussian CUBE, DEN, GRD, GULP, VASP, Voxel, XSF filesVisualize porosity as "negative" (void) or "positive" (filled) space.Calculate and visualize electron density for any crystal.Clamp data display range using range slider.Point cloud (size or opacity) display option.Visualize multiple lattice planes sections, with transparency.Display iso-surfaces or volumes with real-time range control.Flatter your experimental data with bicubic interpolation for gorgeous smooth display.Customize interior, exterior and truncation colours.Choice of colour gradients.Work with multiple datasets in the same window.Add or subtract datasets.
Powerful Measurement
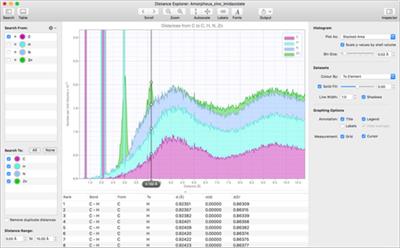
CrystalMaker provides easy-to-use screen tools for measuring coordination environments - bond distances, angles, torsion angles - just by clicking with the mouse, or rolling over different atoms and bonds for real-time output. You can also visualize coordination clusters and shells - with real-time radius adjustment, e.g., build a spherical nanoparticle with a single mouse click. Detailed bond distance and angle information is available on screen, or can be exported to a text file. The powerful Distance Explorer lets you visualize coordination environments graphically, around any combination of elements or sites.
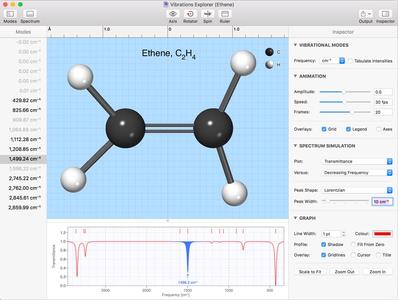
Molecular Modelling and Vibrational Modes
CrystalMaker X features a powerful - and unique - energy-modelling engine that lets you turn crude, hand-drawn molecular structures into lowest-energy three-dimensional structures. Visualize the relaxation in real time - you can even rotate your model as it relaxes. This work is the culmination of a three-year research project in collaboration with world-leading UK scientists. We believe it offers you the best practical solution for molecular modelling - at a fraction of the cost of competitor products.
Why is CrystalMaker's Modelling So Unique?
Force field approach, using parameterised potentials calibrated using DFT: all the advantages of DFT, but works in real time, with much larger systems - and on your laptop!Smart selection of potentials, based on bonding environment: requires no additional user input.Monte Carlo technique allows relaxation of structures far from equilibrium - e.g., crude, hand-drawn structures with excessive bond lengths and distorted bond angles!Hybrid least-squares cycle added to provide greater precision in the closing stages of refinement.Provides energy output, available in graphical form during the course of the refinement cycle, and later, via a menu command.Calculates vibrational modes, including frequencies and simulated infra-red spectrum, plus interative visualizations of vibrations in the Vibrations Explorer.
Molecular Crystal Tools
Our mantra of letting you "see the wood for the trees" is particularly apposite when working with complex molecular crystals. Often there's more to see than just the asymmetric unit - the packing for a start - and other programs struggle with fragmented molecules at the edges of the unit cell.
CrystalMaker X's dedicated tools let you reveal the intact contents of one unit cell, isolate individual molecules or show the asymmetric unit. You can repair "fragmented" molecules, "grow" the Description range along an atom's nearest-neighbours, and make sense out of complexity.
Transformation & Crystal Engineering
CrystalMaker lets you transform the unit cell, changing the lattice type, building a supercell, moving the origin, or applying an arbitrary matrix transformation. You can also project the unit cell onto a chosen lattice plane, to create a "surface cell". Unlike traditional software, CrystalMaker features an elegant, easy-to-use interface, with common choices available as presets. Crystallography without the pain!
CrystalMaker's selection tools let you move, detach, duplicate, hide, delete groups of atoms. Hide or repair molecular fragments - isolate individual molecules. Display lattice planes in any orientation; slice the crystal to investigate surfaces or internal planes - and place one structure or molecule inside another. CrystalMaker's real-time crystal engineering tools let you insert space within your crystal lattice, place new atoms or entire molecules in the crystal - and design new crystal lattices.
Transformation Tools
Description Range: real-time control over atom range along xyzSpherical Cluster: real-time control over inner & outer radii of your "nanocrystal".Cavity Finder: automatically show the largest cavities in your structure using appropriately-sized translucent spheres.Selection Tools (arrow/rectangle, lasso, polygon, zapper) to hide or isolate groups within the structure.Slice lattice with oriented lattice plane: display surface.Slab generation: using two oriented lattice planes.Surface Cell Generation: projected onto a specified plane.Supercell: specify a multiple of the existing cell dimensions.General Matrix Transformation with presets (e.g., convert rhombohedral to hexagonal.)Discard Symmetry command; adds new sites to preserve your structure.Crystal-to-Molecule conversion.Packing Explorer provides interactive molecule-to-crystal command with live symmetry, cell parameters, position and orientation.Move or Rotate Groups of atoms within the crystal lattice.Duplicate Selected Atoms.Change Atom Types (element symbol, occupancy), using the Selection Inspector.Insert Space along a specified lattice plane within the crystal.Bend Lattice: select atoms, specify a radius of curvature and you have an instant nanotube (or bent sheet).Interpolate Structures: add intermediate structures to make animations of structural behaviour smoother.
3D Printing & Model Export
CrystalMaker lets you print 3D models of your structures, via the STL 3D file export option (widely supported by most 3D printers). You can also export any structure as a self-contained COLLADA 3D file: ideal for exchange with 3D graphics software. Even better, you can view your CrystalMaker COLLADA files on your iPad! Just drop your files into iBooks Author (a free download from the Mac app store), then publish to your iPad.
Diffraction Support
Diffraction patterns can be simulated for any displayed crystal structure, with just a single menu command - thanks to our CrystalDiffract (powder diffraction) and SingleCrystal (X-ray, neutron and TEM single-crystal diffraction) programs. SingleCrystal can be linked with CrystalMaker, so as you rotate your structure its diffraction pattern rotates, and vice versa Video. A new "Live Powder Diffraction" mode lets you edit your structure in CrystalMaker and see its corresponding powder diffraction pattern update in real time, in CrystalDiffract.
Buy Premium From My Links To Get Resumable Support,Max Speed & Support Me


